A framework for exhaustively mapping functional missense variants
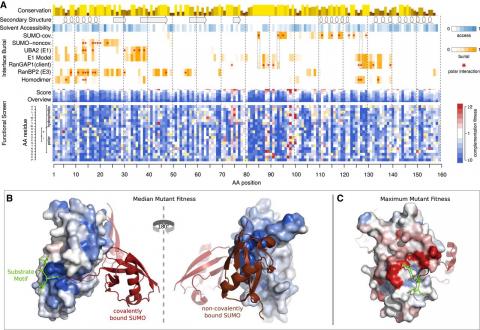
Weile J, Sun S, Cote AG, Knapp J, Verby M, Mellor JC, Wu Y, Pons C, Wong C, van Lieshout N, Yang F, Tasan M, Tan G, Yang S, Fowler DM, Nussbaum R, Bloom JD, Vidal M, Hill DE, Aloy P, Roth FP, Although we now routinely sequence human genomes, we can confidently identify only a fraction of the sequence variants that have a functional impact. Here, we developed a deep mutational scanning framework that produces exhaustive maps for human missense variants by combining random codon mutagenesis and multiplexed functional variation assays with computational imputation and refinement. We applied this framework to four proteins corresponding to six human genes: UBE2I (encoding SUMO E2 conjugase), SUMO1 (small ubiquitin-like modifier), TPK1 (thiamin pyrophosphokinase), and CALM1/2/3 (three genes encoding the protein calmodulin). The resulting maps recapitulate known protein features and confidently identify pathogenic variation. Assays potentially amenable to deep mutational scanning are already available for 57% of human disease genes, suggesting that DMS could ultimately map functional variation for all human disease genes.
Molecular Systems Biology,
2017, 13(12), 957
Pubmed: 29269382
Direct link: 10.15252/msb.20177908