Research in our lab mainly focus on understanding the molecular bases of how macromolecular complexes and cell networks operate by analyzing protein-protein interaction networks with the help of high-resolution 3D structures.Proteins are the main perpetrators of most cellular tasks. However, they seldom act alone and most biological processes are carried out by macromolecular assemblies and regulated through a complex network of protein-protein interactions. Thus, modern molecular and cell biology no longer focus on single macromolecules but now look into complexes, pathways or even entire organism interactomes. The many genome-sequencing initiatives have provided a near complete list of the components present in an organism, and post-genomic projects have aimed to catalog the relationships between them. The emerging field of systems biology is now mainly centered on unraveling these relationships and trying to use the information that they contain to boost novel biomedical applications. Accordingly, in the Structural Bioinformatics & Network Biology group (SB&NB), we have established two interrelated research lines devoted to reveal the molecular bases of how macromolecular complexes and cell networks operate.
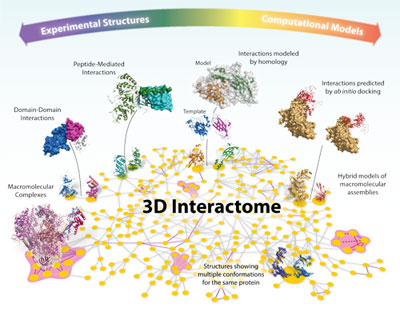
Pushing structural details into large molecular machines and interactome networks
The emergence of high-throughput proteomic initiatives devoted to the identification of new protein interactions and macromolecular complexes has led to the production of large interactome maps for several model organisms, including human. All these interaction maps lack in molecular details: they tell us who interacts with whom, but not how. A full understanding of how molecules interact can be attained only from high resolution three-dimensional (3D) structures, since these provide crucial atomic details about binding. Unfortunately, only a few among the discovered complexes and interactions in high-throughput initiatives meet the high-quality standards required to be promptly used in structural studies, which has created an increasing gap between the number of known protein interactions and complexes and those for which a 3D structure is available. In our group, we have developed and validated a computational strategy to identify those complexes found in high-throughput affinity purification experiments that will stand the best chances to successfully express, purify and crystallize with little further intervention (Pache & Aloy, 2008, Proteomics). Furthermore, we used all the available computational techniques to try to enlarge the structural coverage of entire interactomes. In this context, we ran the first-ever high-throughput protein-protein docking experiment, applying state-of-the-art docking procedures to 217 experimental structures and 1,023 homology models, providing structural models for over 3,000 protein-protein interactions in the yeast interactome (Mosca et al. 2009, PLoS Comp Biol). Another area in which we have been intensively working is that of peptide-mediated protein interactions, where a globular domain in one protein recognizes a linear peptide from another, creating a relatively small interface. In the lab, we have systematically identified all instances of peptide-mediated protein interactions of known three-dimensional structure and used them to investigate the individual contribution of motif and context to the global binding energy. Our analysis partially revealed the molecular mechanisms responsible for the dynamic nature of peptide-mediated interactions, and suggested a global evolutionary mechanism to maximise the binding specificity (Stein & Aloy, 2008, PLoS ONE). Furthermore, we have identified particular structural features of these interactions, and used them to identify several novel groups of peptide-domain pairs in 3D structures (Stein & Aloy, 2010, PLoS Comp Biol). They are available to the scientific community in our database 3did, which provides high-resolution 3D structures of modular protein interactions between pairs of globular domains as well as domains and peptides (Stein et al., NAR 2011).
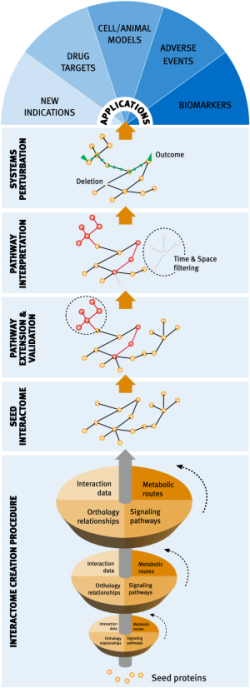
Network medicine approaches to complex diseases
Network and systems biology offer a novel way of approaching drug discovery by developing models that consider the global physiological environment of protein targets, and the effects derived of tinkering with them, without losing the key molecular details. In particular, we claim that network biology will play a central role in the development of novel polypharmacology strategies to fight complex multi-factorial diseases, where efficacious therapies will need to centre on bringing down entire pathways rather than single proteins. Accordingly, the main goal of our laboratory is the global molecular characterization of pathological pathways through a combination of computational biology and interaction discovery techniques, in a real dry-wet cycle, where we use computational modelling to design the experiments needed to complement and complete the initial models.
To this end, the laboratory takes advantage of the Experimental Bioinformatics Lab, which gives us the possibility of carrying out the molecular/cell biology experiments necessary to complete and validate our in silico models and hypothesis.
Research directions that has been active in this line has been:
- the development of computational tools to build, analyze and extract biologically relevant information from interactome networks (Pujol et al. 2010. TiPS; Pache & Aloy, 2010, P Natl Acad Sci, under review). In particular, we have developed methods to assess the completeness and reliability of protein interaction networks, as well as determining the most accurate/noisy areas in the interaction maps and select the optimal points (i.e. proteins) to improve them.
- exploratory projects to extend the interactome networks associated to the canonical pathways closely related to the onset and development of Alzheimer´s disease (AD) (Soler-López et al. 2010. Genome Res, In Press), colorectal cancer (CRC), breast cancer (BC).
- the identification of novel substrates for the oncogene Aurora A, a S/T kinase essential for cell cycle progression. In this case, we have developed a computational approach that integrates distinct types of biological information to generate a ranked list of 90 potential substrates, with a predicted accuracy, determined by experimental validation on a randomly selected group of candidates, of about 80%. (Sardón et al. 2010. EMBO Rep).
Finally, collaboration with several pharmaceutical and biotechnological companies is ongoing in order to use the compound-target data that they have gathered over the last 30 years as a means to simulate perturbations in key points of the networks by means of chemical intervention (i.e. drug modulation) to convert static and noisy interation maps into mathematical models, able to reproduce phenotypic observations.