Pushing structural information into the yeast interactome by high-throughput protein docking experiments
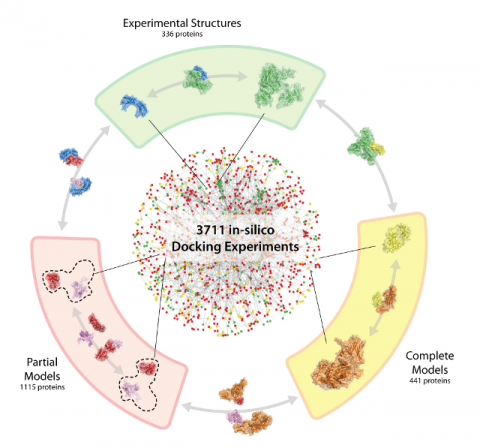
Mosca R, Pons C, Fernández-Recio J, Aloy P, The last several years have seen the consolidation of high-throughput proteomics initiatives to identify and characterize protein interactions and macromolecular complexes in model organisms. In particular, more that 10,000 high-confidence protein-protein interactions have been described between the roughly 6,000 proteins encoded in the budding yeast genome (Saccharomyces cerevisiae). However, unfortunately, high-resolution three-dimensional structures are only available for less than one hundred of these interacting pairs. Here, we expand this structural information on yeast protein interactions by running the first-ever high-throughput docking experiment with some of the best state-of-the-art methodologies, according to our benchmarks. To increase the coverage of the interaction space, we also explore the possibility of using homology models of varying quality in the docking experiments, instead of experimental structures, and assess how it would affect the global performance of the methods. In total, we have applied the docking procedure to 217 experimental structures and 1,023 homology models, providing putative structural models for over 3,000 protein-protein interactions in the yeast interactome. Finally, we analyze in detail the structural models obtained for the interaction between SAM1-anthranilate synthase complex and the MET30-RNA polymerase III to illustrate how our predictions can be straightforwardly used by the scientific community. The results of our experiment will be integrated into the general 3D-Repertoire pipeline, a European initiative to solve the structures of as many as possible protein complexes in yeast at the best possible resolution. All docking results are available at http://gatealoy.pcb.ub.es/HT_docking/.
PLoS computational biology,
2009, 5(8), e1000490
Pubmed: 19714207
Direct link: 10.1371/journal.pcbi.1000490
Link to the supplementary material